转录组分析(上游+下游)
比较流行的电脑系统介绍:
操作系统(Operating System)
├── Windows
├── macOS
└── Linux(内核)
│
├── Debian(基础发行版)
│ ├── Ubuntu(桌面/科研开发)
│ └── Raspberry Pi OS(树莓派)
│
└── CentOS(企业服务器,传统主流发行版)
服务器连接:
ssh安全远程登录协议+FTP文件传输协议
你的电脑
│
│ SSH协议(加密)
│========================>
│
树莓派(SSH Server)
你的电脑
│
│ FTP协议
▼
FTP服务器
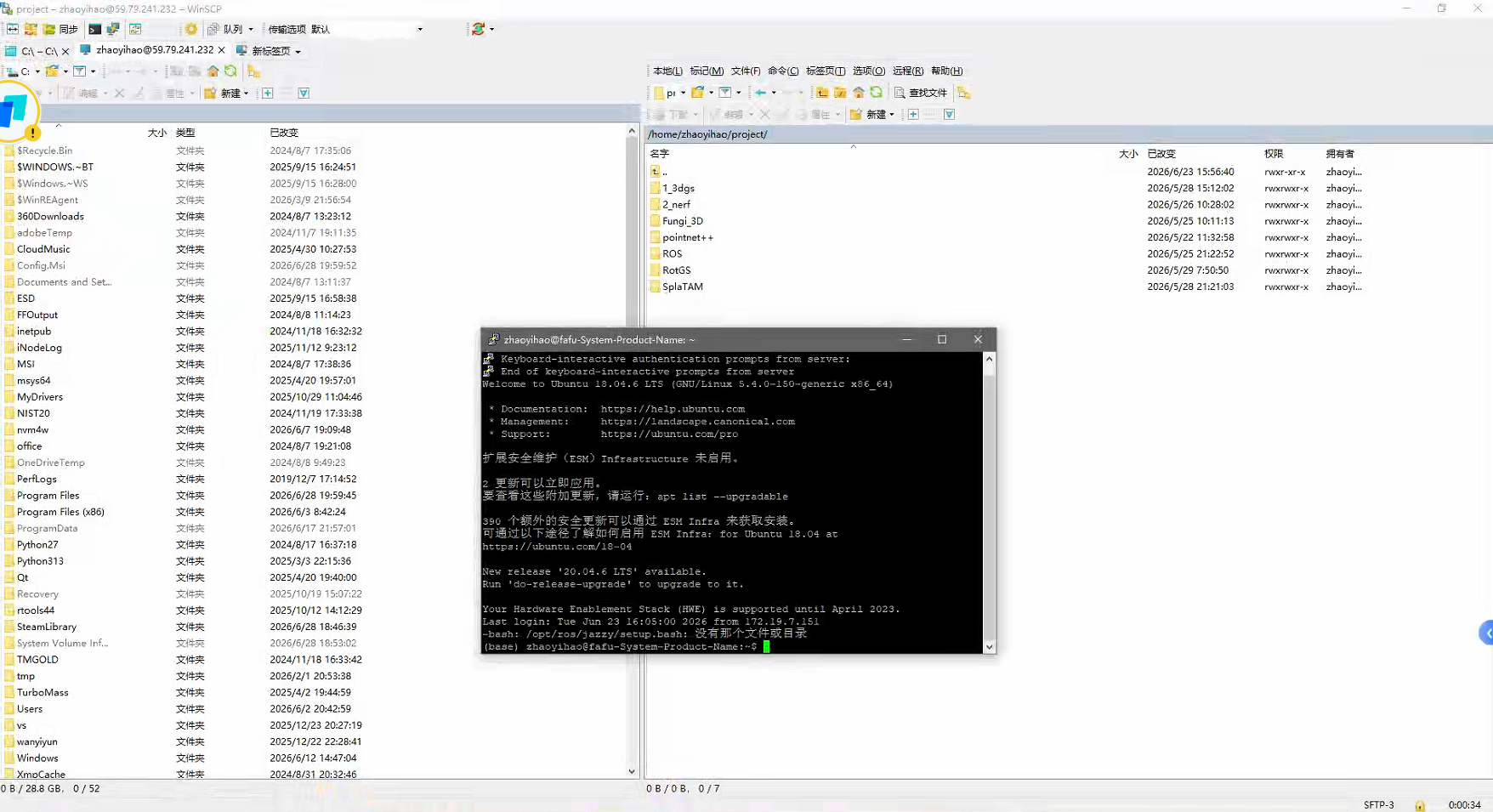
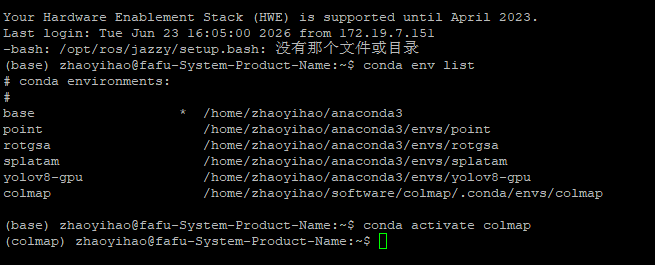
1.软件安装
#直接在服务器中进行安装即可:
#首先是环境管理工具:conda
1.质控:fastqc, multiqc, cutadapt, trim-galore
2.序列比对:hisat2
3.表达定量:subreadconda安装
wget -c https://repo.anaconda.com/archive/Anaconda3-2023.03-1-Linux-x86_64.sh
bash Anaconda3-2023.03-1-Linux-x86_64.sh
echo 'export PATH=~/anaconda3/bin:$PATH' >> ~/.bashrc
source ~/.bashrc
2.数据准备
#原始测序数据(下机数据集)
fq.gz文件格式的数据
#参考基因组数据
1.全基因组数据(fasta文件)
2.全基因组注释文件(gff3/gtf数据,用gff3数据最好,因为gff3可以转gtf文件。反之不行)
3.全基因组蛋白序列文件(做功能注释使用)原始数据准备:
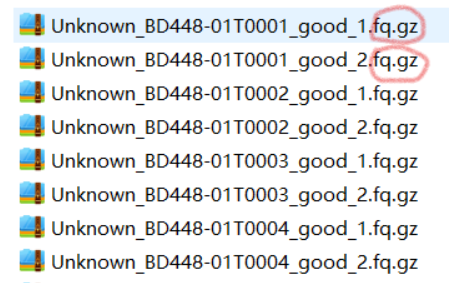
双端测序数据如上。
参考基因组准备
需要数据:
fasta文件结构:
>基因id
ATCG/一堆氨基酸序列
>基因id
ATCG/一堆氨基酸序列gff文件结构
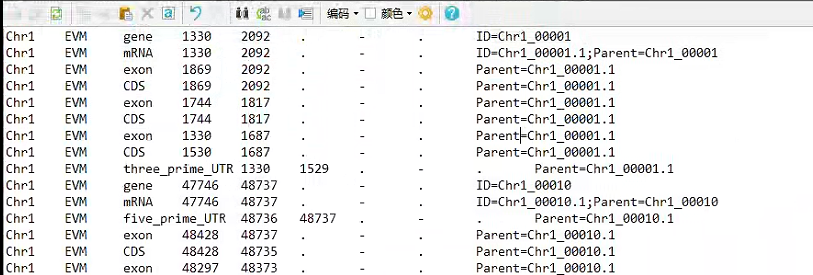
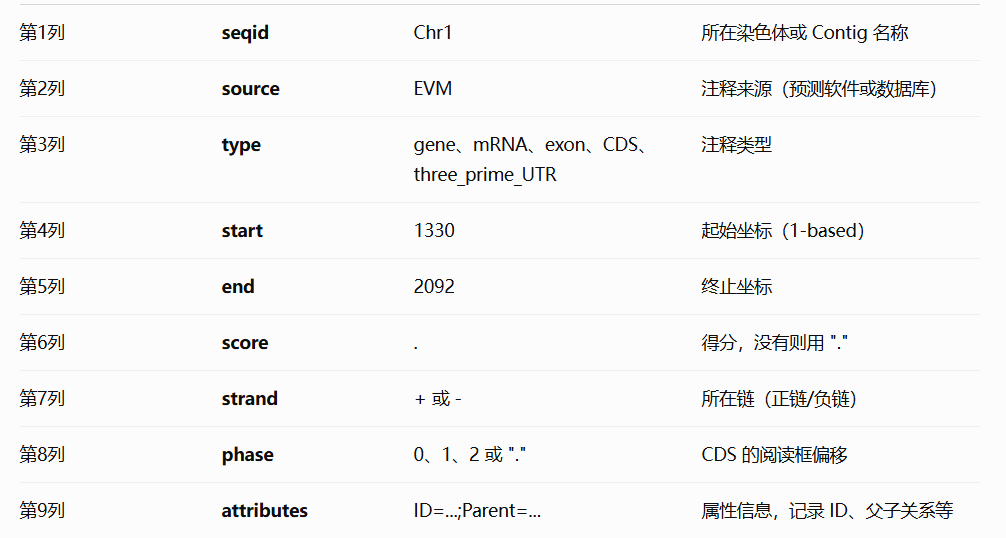
seqid :参考序列的id。
source:注释的来源。如果未知,则用点(.)代替。一般指明产生此gff3文件的软件或方法。
type: 类型,此处的名词是相对自由的,建议使用符合SO惯例的名称(sequenceontology),如gene,repeat_region,exon,CDS等。
start:开始位点,从1开始计数(区别于bed文件从0开始计数)。
end:结束位点。
score:得分,对于一些可以量化的属性,可以在此设置一个数值以表示程度的不同。如果为空,用点(.)代替。
strand:“+”表示正链,“-”表示负链,“.”表示不需要指定正负链。
phase :步进。对于编码蛋白质的CDS来说,本列指定下一个密码子开始的位置。可以是0、1或2,表示到达下一个密码子需要跳过的碱基个数。
attributes:属性。一个包含众多属性的列表,格式为“标签=值”(tag=value),不同属性之间以分号相隔。
下列的标签已定义:
ID:指定一个唯一的标识。对属性分类是非常好用(例如查找一个转录单位中所以的外显子)。
Name:指定属性的名称。展示给用户的就是该属性。
Alias:名称的代称或其它。当存在其它名称时使用该属性。
Note:描述性的一些说明。
Alias和Note可以有多个值,不同值之间以逗号分隔。
如:Alias=M19211,gna-12,GAMMA-GLOBULIN
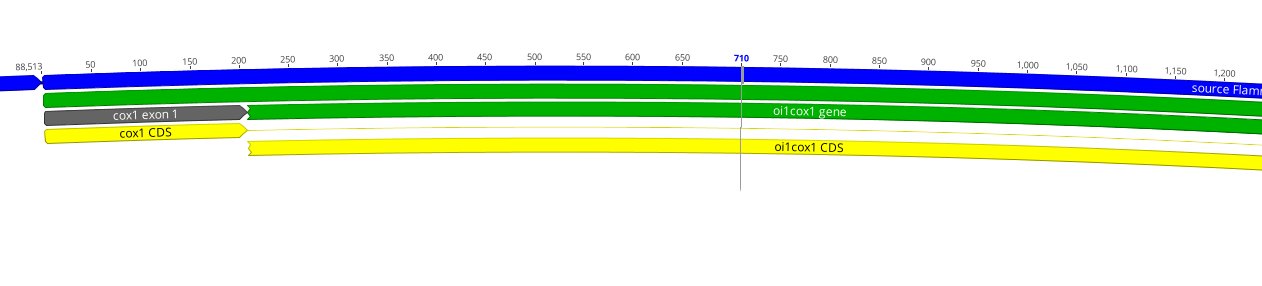
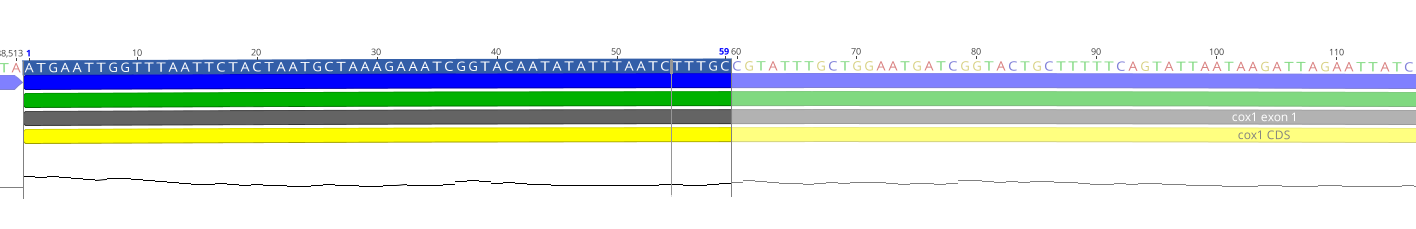
可视化举例:
如下是gff(注释文件)和fasta文件结合到一块去看的一个软件:标注的有具体的信息和序列。
3.质控和过滤
质控:
这个一般是测序公司干的,我这边简要带过一下
fastqc *fq.gz
#运行后会直接出:
html文件和zip文件
#也就是质控报告。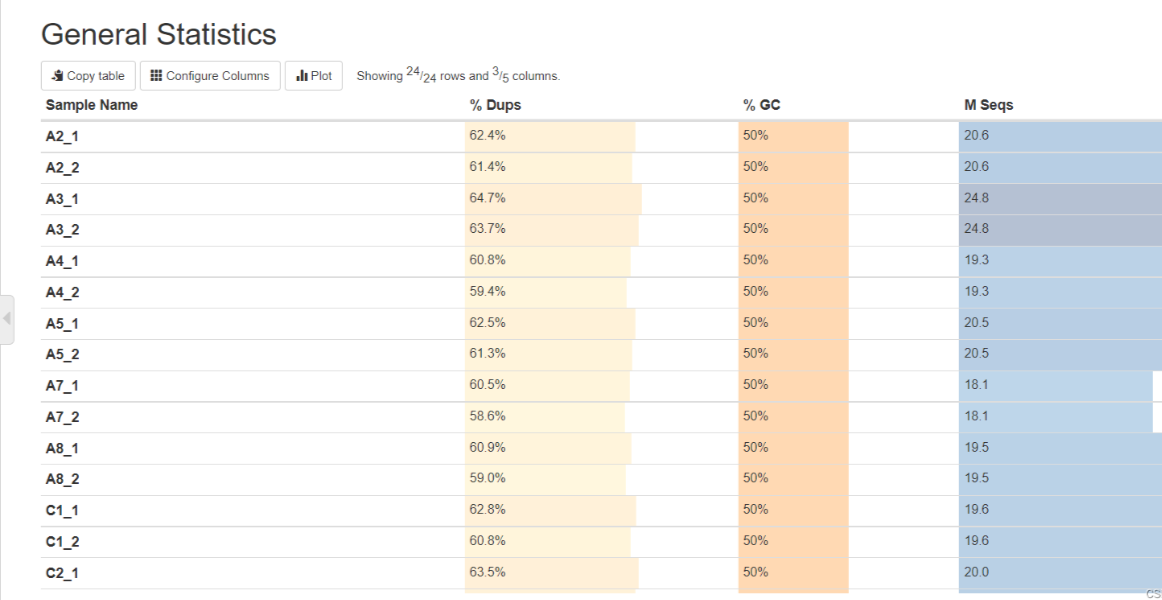
General Statistics——所有样本数据基本情况统计
%Dups——重复reads的比例,比例越高代表重复reads数量就越多,有用的reads就少。
%GC——GC含量占总碱基的比例,比例越小越好
M Seqs——总测序量(单位:millions)
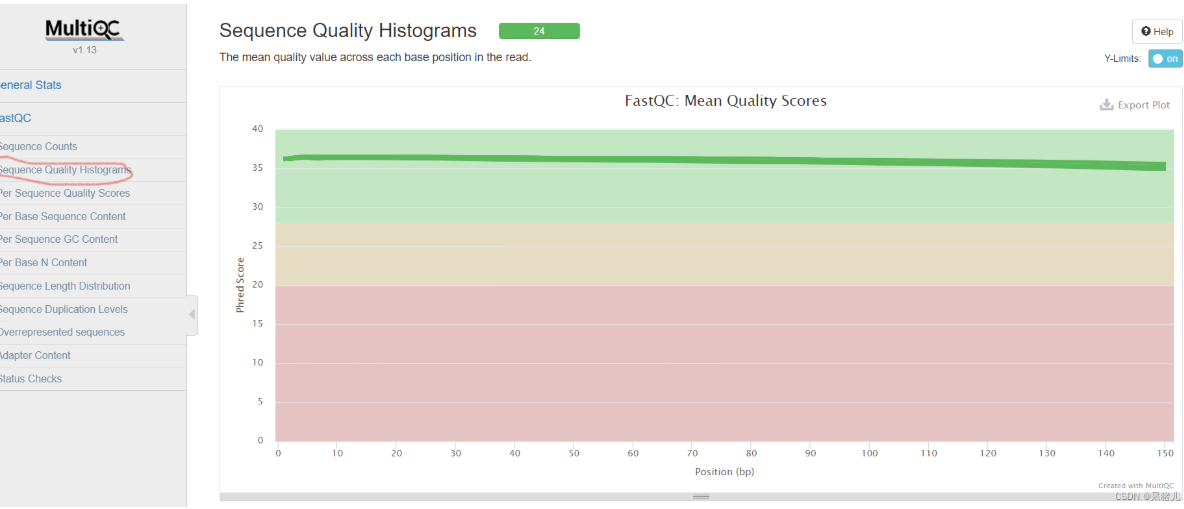
绿色的表示质量可以。多余的没有了解的必要,如果没达到要求,直接让测序公司回去重新测即可。
过滤
trim_galore -help
--quality:设定Phred quality score阈值,默认为20,表示质量分数小于20的会被舍弃,分析时可改成25,稍微严格一些。
--stringency:设定可以忍受的前后adapter重叠的碱基数,默认为1(非常苛刻),值越大越宽松,可以根据实验需求适度放宽。
--length:设定输出reads长度阈值,就是长度不达标的reads会被抛弃。
--paired:对于双端测序结果,一对reads中,如果有一个被剔除,那么另一个会被同样抛弃,而不管是否达到标准。
--output_dir:输出目录,就是过滤后的文件输出的路径(需要提前建立目录,否则运行会报错)。
#过滤代码:
trim_galore --quality 25 --stringency 1 --length 50 --paired --output_dir /home/daizhuer/Desktop/02_clean_data/ A2_1.fq.gz A2_2.fq.gz
--quality:25 代表质量小于25会被剔除
--stringency 1 代表对接头种重叠碱基数检查非常严格,如果接头中重叠碱基数大于1在后面的分析报告中就会显示出来
--length 设定的是50,表示reads长度小于50的会被舍弃
--paired 双端测序
--output_dir 输出路径
然后后面跟的是两个输入文件。4.序列比对
软件:hisat2,samtools构建参考基因组索引:hisat2-build进行构建:
hisat2-build ref.fasta ./01_ref/genome这个步骤比较慢
构建索引这个步骤就是说将对应序列的位置给出来,构建索引,比对就不需要去每个序列重新比对了。
hisat2进行比对,生成sam文件
hisat2 --new-summary -p 2 -x ./01_ref/genome -1 ./02_clean_data/A2_1_val_1.fq.gz -2 ./02_clean_data/A2_2_val_2.fq.gz -S ./03_hisat2_result/A2.sam --rna-strandness RF
--new-summary #输出新版统计信息。
-p 2 #表示线程
--rna-strandness RF #文库是否具有链特异性(Strand-specific)
RF这个就是文库方向
RF表示,比对是从左往右比对,
FR表示, 比对是从右往左比对。
文库方向确定
RSeQC进行文库方向确定(如果是链特异性文库的话,如果是非链特异性文库就不用加这个参数)
Reading reference gene model ...
This is PairEnd Data
Fraction of reads failed to determine: 0.02
Fraction of reads explained by "1++,1--,2+-,2-+": 0.95
Fraction of reads explained by "1+-,1-+,2++,2--": 0.03
#这个95%表示选择RF
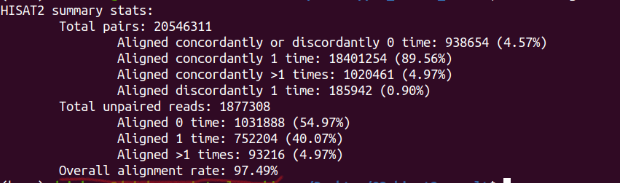
根据比对率选择参考基因。
压缩排序sam文件---bam文件,相当于sam文件的压缩包
samtools sort -o ./03_hisat2_result/A2.bam ./03_hisat2_result/A2.sam
#sam_bam:二进制压缩
#按照基因组坐标排序(Sort)(按照参考进行排列)5.表达定量
使用featureCounts软件进行定量比对
featureCounts -a ./01_ref/Mus_musculus.GRCm39.108.gtf -T 3 -p --countReadPairs -g gene_id -t exon -M -O -o ./04_quantify/A2 ./03_hisat2_result/A2.bam
#-a后面跟的gtf文件
#-T线程
#-p 是在双端测序的情况下使用,会统计fragment而不是read
#-g gene_id基因表达,肯定用这个。
# -t exon 指定定量的类型,默认是exon(外显子),这就是说当read落到外显子上才会被统计
#-M 多重read会被统计
#-O 允许多重比对,就是说当一个read比对到多个外显子时,这条read会被统计多次
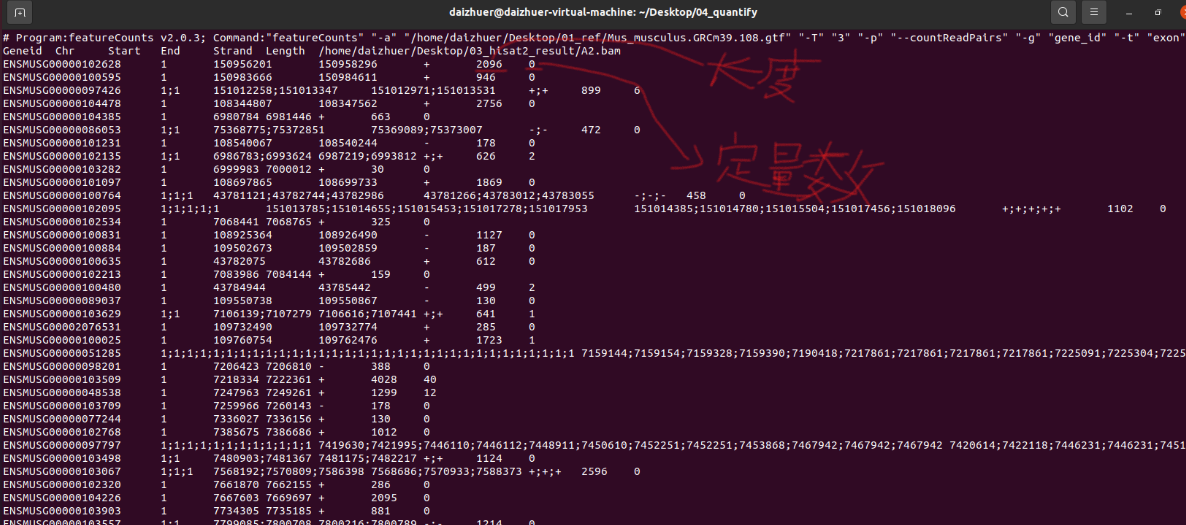
如上就是单个的定量的最后结果。
Geneid 是基因id(重点关注)
Chr 是该基因位于哪条染色体上
Start 表示该基因起始位置
End 表示该基因结束位置
Strand 表示正负链
Length 基因长度,后面计算RPKM和TPM会用到(重点关注)
最后一列就是定量的结果(就是定量到该基因上的reads有多少条)(重点关注)
最后提取第一列和第七列,也就是id信息和counts信息
并合并多个样本的counts数据和信息。
按照我们的就是:
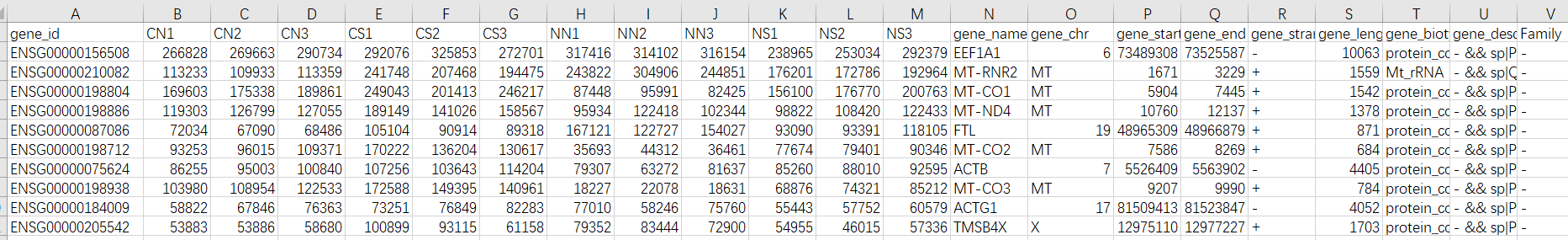
转录组下游分析
主要涉及了差异基因的筛选和各种图的绘制,这个图应该都看得懂。
DESeq2: 需要原始的count矩阵(如通过htseq-count工具获得),并且只支持重复样品。
limma: 可以接受原始的count矩阵,但需要用户自行进行标准化(通常是log 转换),并且也支持重复样品。
edgeR: 要求输入原始的count矩阵,既支持单个样品也支持重复样品。
也就是如上三种方法,对应三个R包。
1.5 在做转录组分析的时候该选用哪种方法?(关键)
GEO芯片数据——用limma差异分析,因为GEO芯片数据底层已经对数据做了标准化处理。
(数据参考:——数据处理(GEO数据库——芯片数据))
GEO高通量数据——用DESeq2差异分析,因为可以获取到count数据。
(数据参考:——数据处理(GEO数据库——高通量测序数据))
TCGA_count数据——用DESeq2差异分析,因为可以获取到count数据。
(数据参考:——数据处理(TCGA数据库))
自测序数据——用DESeq2差异分析,因为可以获取到count数据。
然后目前用到最多的就是DESeq2这个R包进行的。
library(DESeq2)
其他一些做数据整理的包这边就不提及了。差异基因的筛选
log2FoldChange = log2(前一组表达量 / 后一组表达量).
这个表达量就是针对DeSeq2自动对counts进行标准化的一个结果。
log2FoldChange
1.计算每个基因的几何平均数
所有数先相乘,再开 n 次方
2.计算ratio
每个样本的 Count /这个基因的几何平均
3.取中位数
Size Factor
4.标准化
然后用每个count/SizeFactor即可。
之后计算log2FoldChange,进行比对。
Padj
负二项分布(Negative Binomial)模型:
Padj(FDR校正)
表达量获取等等:CPM、FPKM和TPM表达量的获取
CPM
cpm <- function(counts){
lib.size <- colSums(counts)
sweep(counts, 2, lib.size, "/") * 1e6
}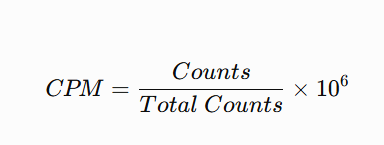
FPKM
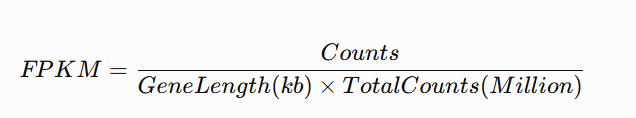
fpkm <- function(counts, gene_length){
lib.size <- colSums(counts) / 1e6
gene.length.kb <- gene_length / 1000
t( t(counts) / gene.length.kb ) / lib.size
}TPM
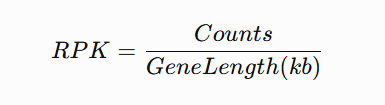
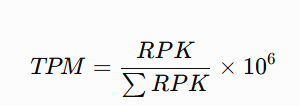
tpm <- function(counts, gene_length){
length.kb <- gene_length / 1000
rpk <- sweep(counts, 1, length.kb, "/")
sweep(rpk, 2, colSums(rpk), "/") * 1e6
}Raw Counts
│
├── CPM(只校正测序深度)
│
├── FPKM(校正长度 + 深度)
│
└── TPM(先长度,再统一比例 ★推荐)
GO富集和KEGG富集
先拿蛋白序列文件做功能注释使用。
eggnogEggNOG Database | Orthology predictions and functional annnotaion
本地注释也可以,最终得到一个表:
各个数据库的富集分析,得到各个基因对应在数据库中的功能。
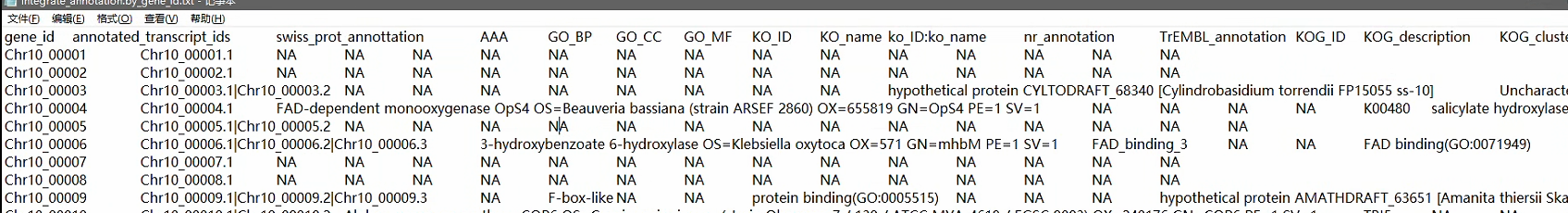
去vlookup到差异基因的列中,之后进行绘图。
转录组下游分析图
1.箱线图(盒形图)
横轴就是分组嘛,纵轴是log2(FPKM+1),先进行初步计算,然后绘制箱线图即可。
核心代码:按需更改
# 加载必要的包
library(readxl)
library(ggplot2)
library(tidyr)
# 读取Excel数据(请修改为您的文件路径)
df <- read_excel("your_data.xlsx")
# 数据重塑:将宽格式转换为长格式
# 假设您的数据列名为:group, FPKM, value2, value3, value4
df_long <- df %>%
pivot_longer(cols = c(FPKM, value2, value3, value4),
names_to = "variable",
values_to = "value")
# ============ 核心绘图代码 ============
# 方法1:按原始分组绘制箱线图(以value4为例)
ggplot(df, aes(x = group, y = value4)) +
geom_boxplot(fill = "steelblue", alpha = 0.7) +
geom_jitter(width = 0.2, size = 1.5, alpha = 0.5) +
theme_minimal() +
labs(title = "FPKM Expression Distribution",
x = "Group",
y = "FPKM Value")
# 方法2:按主要分组(CN/CS/NN/NS)绘制箱线图
df$main_group <- substr(df$group, 1, 2) # 提取前两个字符作为主分组
ggplot(df, aes(x = main_group, y = value4, fill = main_group)) +
geom_boxplot(alpha = 0.7, outlier.color = "red", outlier.size = 2) +
geom_jitter(width = 0.2, size = 1.5, alpha = 0.4) +
scale_fill_brewer(palette = "Set2") +
theme_classic() +
labs(title = "FPKM Expression by Group",
x = "Group",
y = "FPKM Value") +
theme(legend.position = "none")
# 方法3:绘制所有变量的箱线图(如果有多组测量值)
df_long %>%
ggplot(aes(x = group, y = value, fill = variable)) +
geom_boxplot(alpha = 0.7, position = position_dodge(0.8)) +
theme_minimal() +
labs(title = "Multiple Measurements Boxplot",
x = "Group",
y = "Value") +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
# 方法4:带统计标记的进阶版本
ggplot(df, aes(x = main_group, y = value4, fill = main_group)) +
geom_boxplot(alpha = 0.7, width = 0.6) +
geom_jitter(width = 0.15, size = 2, alpha = 0.6, color = "black") +
stat_summary(fun = mean, geom = "point", shape = 23, size = 3, fill = "red") +
stat_summary(fun.data = mean_se, geom = "errorbar", width = 0.2) +
scale_fill_manual(values = c("#66C2A5", "#FC8D62", "#8DA0CB", "#E78AC3")) +
theme_bw() +
labs(title = "FPKM Boxplot with Statistics",
x = "Treatment Group",
y = "FPKM Expression") +
theme(legend.position = "none",
plot.title = element_text(hjust = 0.5, size = 14),
axis.text = element_text(size = 11),
axis.title = element_text(size = 12))2.FPKM密度图
# 模拟FPKM数据
set.seed(123)
fpkm_values <- rnorm(10000, mean = 1, sd = 0.5) # log10(FPKM+1) 值
fpkm_values <- pmax(fpkm_values, 0) # 确保非负
# 方法1:手动计算密度
hist_info <- hist(fpkm_values, breaks = 50, plot = FALSE)
manual_density <- hist_info$counts / (length(fpkm_values) * diff(hist_info$breaks[1:2]))
print(head(manual_density))
# 方法2:使用内置密度函数
density_obj <- density(fpkm_values)
print(head(density_obj$y)) # 这就是纵轴的密度值
# 可视化对比
par(mfrow = c(1, 2))
# 直方图(纵轴为频率)
hist(fpkm_values, breaks = 50, main = "Frequency Histogram",
xlab = "log10(FPKM+1)", ylab = "Frequency")
# 密度图(纵轴为密度)
hist(fpkm_values, breaks = 50, main = "Density Plot",
xlab = "log10(FPKM+1)", ylab = "Density", freq = FALSE)
lines(density(fpkm_values), col = "red", lwd = 2)
# 验证:曲线下面积 ≈ 1
area_under_curve <- sum(density_obj$y) * diff(density_obj$x[1:2])
print(paste("曲线下面积 =", area_under_curve)) # 应该接近 13相关性热图
library(ggplot2)
library(reshape2)
library(readxl)
library(scales)
# ============ 方法1:使用corrplot包(最推荐)============
library(corrplot)
# 读取数据(假设您的数据是FPKM矩阵,行=基因,列=样本)
df <- read_excel("your_fpkm_data.xlsx")
# 假设第1列是基因名,其余列是样本
fpkm_matrix <- as.matrix(df[, -1])
rownames(fpkm_matrix) <- df[[1]]
# 计算样本间相关性(Pearson相关系数)
cor_matrix <- cor(fpkm_matrix, method = "pearson")
# 绘制热图
corrplot(cor_matrix,
method = "color", # 颜色填充
type = "upper", # 显示上三角
tl.col = "black", # 标签颜色
tl.srt = 45, # 标签旋转角度
col = colorRampPalette(c("blue", "white", "red"))(100),
addCoef.col = "black", # 显示相关系数
number.cex = 0.7, # 数字大小
title = "Sample Correlation Matrix",
mar = c(0, 0, 2, 0))
# ============ 方法2:使用ggplot2(更灵活)============
library(ggplot2)
library(reshape2)
# 将相关矩阵转为长格式
cor_long <- melt(cor_matrix)
# 自定义颜色(匹配您的图:红-白-蓝)
ggplot(cor_long, aes(Var1, Var2, fill = value)) +
geom_tile(color = "white", linewidth = 0.5) +
scale_fill_gradient2(
low = "blue",
mid = "white",
high = "red",
midpoint = 0.95,
limits = c(0.9, 1.0),
oob = squish
) +
geom_text(aes(label = round(value, 2)),
color = "black",
size = 3,
fontface = "bold") +
labs(
title = "Sample Correlation Matrix",
x = "",
y = "",
fill = "Correlation"
) +
theme_minimal() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1, size = 10),
axis.text.y = element_text(size = 10),
panel.grid = element_blank(),
plot.title = element_text(hjust = 0.5, size = 14, face = "bold"),
legend.position = "right"
)
# ============ 方法3:带聚类和分组注释的进阶版 ============
library(pheatmap)
# 创建分组注释
annotation_col <- data.frame(
Group = factor(substr(colnames(cor_matrix), 1, 2))
)
rownames(annotation_col) <- colnames(cor_matrix)
# 定义分组颜色
ann_colors <- list(
Group = c(CN = "#E41A1C", CS = "#377EB8",
NN = "#4DAF4A", NS = "#984EA3")
)
# 绘制热图(带聚类和注释)
pheatmap(cor_matrix,
color = colorRampPalette(c("blue", "white", "red"))(100),
breaks = seq(0.9, 1.0, length.out = 101),
display_numbers = TRUE,
number_color = "black",
number_format = "%.2f",
fontsize_number = 8,
annotation_col = annotation_col,
annotation_colors = ann_colors,
clustering_distance_rows = "euclidean",
clustering_distance_cols = "euclidean",
main = "Sample Correlation Heatmap",
border_color = "white",
cellwidth = 35,
cellheight = 35)
# ============ 方法4:保存高质量PDF ============
pdf("sample_correlation_heatmap.pdf", width = 10, height = 8)
pheatmap(cor_matrix,
color = colorRampPalette(c("blue", "white", "red"))(100),
display_numbers = TRUE,
number_color = "black",
number_format = "%.2f",
fontsize_number = 8,
annotation_col = annotation_col,
annotation_colors = ann_colors,
main = "Sample Correlation Heatmap")
dev.off()
openEuler 是由开放原子开源基金会孵化的全场景开源操作系统项目,面向数字基础设施四大核心场景(服务器、云计算、边缘计算、嵌入式),全面支持 ARM、x86、RISC-V、loongArch、PowerPC、SW-64 等多样性计算架构
更多推荐

 1
1 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)